
Paul Thornalley’s blog – insights from deliberations, investigations and reflections
Hello. This is the start of a blog by Paul J Thornalley. I would like to discuss the simple principle of unscheduled glycolysis and glycolytic overload that underlies the pathogenesis of high glucose concentration in insulin resistance, beta-cell glucotoxicity, development of type 2 diabetes and vascular complications of diabetes and the impaired incretin effect. I then discuss how this can be alleviated by novel targeted therapy.
Pathogenesis of hyperglycemia mediated through glycolytic overload and its alleviation by precision dietary supplement, GlucoRegulate
The damaging effects of hyperglycemia are a consequence of hexokinase-gated increased flux of glucose metabolism without increased glycolytic enzyme activities to accommodate it. We will explore what these means in a moment but overlooking the key role of hexokinases in controlling flux of glucose metabolism for the last 25 years has impaired advances in understanding of the pathogenesis of hyperglycemia and its alleviation. So, let’s see what’s been missing.
Normally when there is increased flux of glucose metabolism into cells – such as skeletal muscle cells in the absorptive period, the activities of glycolytic enzymes downstream of hexokinases 1 and 2 are increased such that steady-state levels of glycolytic intermediates are kept within tolerable levels. It is when increased flux of glucose metabolism – initiated by stabilization of hexokinase-2 to proteolysis by high cellular glucose concentration at sites of vascular complications, for example – occurs without increased activities of glycolytic enzymes downstream that glycolytic intermediates increase to levels that activate multiple pathways of pathogenesis. We described this in our paper of last year in detail – Hexokinase-linked glycolytic overload and unscheduled glycolysis in hyperglycemia-induced pathogenesis of insulin resistance, beta-cell glucotoxicity, and diabetic vascular complications, Rabbani & Thornalley, Frontiers in Endocrinology, 14: 1268308, 2024.
The difference between normal glucose metabolism and glycolytic overload can be likened to that of a dam holding back water with minimal overflow (normal glucose metabolism).

In contrast, glycolytic overload may be likened to where the dam is breached by excessive water input, overtopping the dam wall and causing flooding into expanded areas with resulting damage through excessive water going where it normally doesn’t.

A visual analogy of glycolytic overload.
Paul J Thornalley 12th August 2025
Hexokinases 1 and 2 – normally protective against damaging effects of high glucose concentration but protection is subverted for hexokinase-2 in prolonged hyperglycemia
Hexokinase-1 (HK1) has KM(Glucose) of 61 μM and Hexokinase-2 (HK2) has KM(Glucose) = 340 μM, so they are normally highly saturated with glucose under physiological conditions. This means in the absorptive phase (after a meal), blood glucose concentration increases but the rate of glucose metabolism in tissues where HK1 and HK2 control entry of glucose into metabolism stays the same. This protects against glycolytic overload the brain and heart – mostly HK1, and vascular tissues – HK1 and HK2 (including kidney retina and peripheral nerve). It also means there is glucose ready and waiting to be metabolized in these tissues. So, no need to worry about neurons of the brain running short on glucose!
When there is a prolonged period of increased cellular glucose, something happens to HK2. It is stabilized to proteolysis and thereby HK2 protein and activity increase without other increase of other glycolytic enzymes. The half-life of HK2 is normally 8 – 10 h and approximately doubles when exposed to high glucose concentration. This increases the concentration of HK2 approximately 2-fold. Where HK2 contributes significantly to total hexokinase activity, it increase flux of glucose metabolism by 2 – 3 fold without increase in activity of other glycolytic enzymes, initiating glycolytic overload. Where is HK2 a major player in hexokinase activity? Vascular cells, renal cells, retina, Schwann cells of peripheral nerve and early-stage embryo – all vulnerable to glycolytic overload in prolonged periods of high hyperglycemia. HK2-linked glycolytic overload is now seen as a key initiator of hyperglycemia-induced pathogenesis in diabetic vascular disease – including endothelial dysfunction and increase risk of coronary artery disease, diabetic kidney disease, diabetic retinopathy, diabetic neuropathy and diabetic embryopathy (Rabbani & Thornalley, Frontiers in Endocrinology, 14: 1268308, 2024).
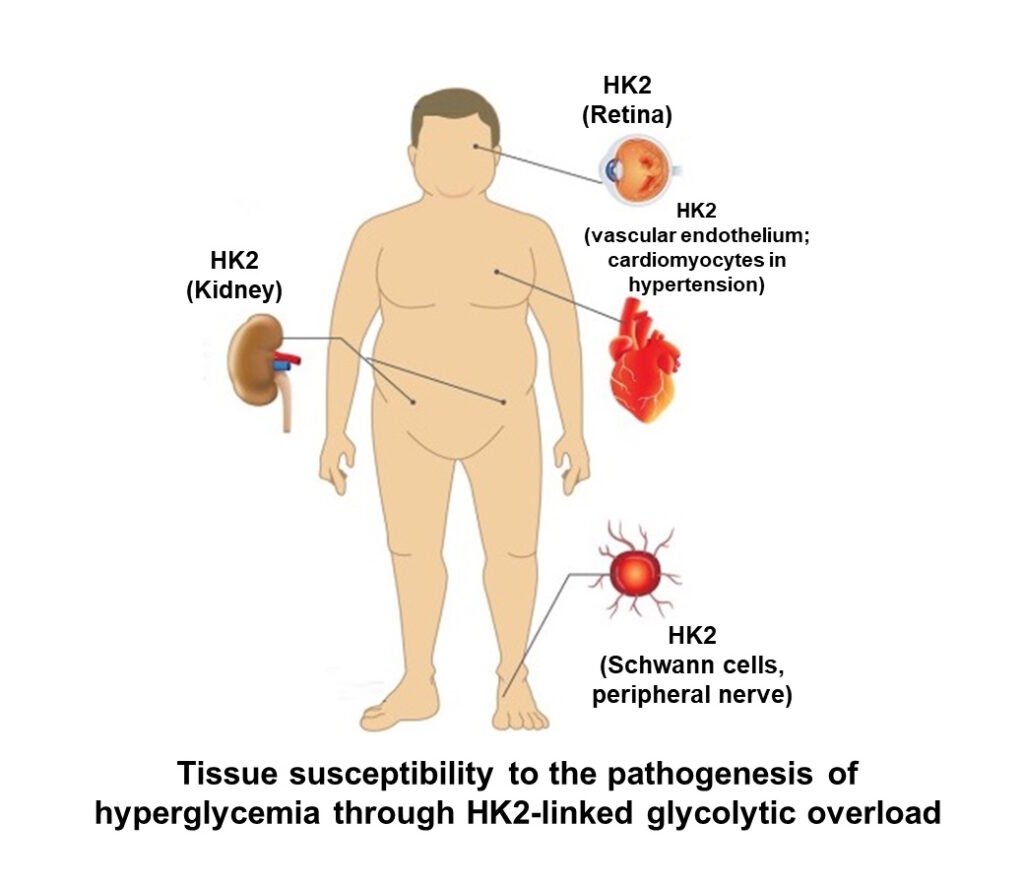
Paul J Thornalley 13th August 2025
Unscheduled glycolysis in peripheral insulin resistance
Hexokinase-2 is also a major hexokinase in skeletal muscle and adipose tissue. These are sites where glucose is taken up at increased rates in the absorptive phase by insulin-stimulated recruitment of GLUT4 glucose transporters to the plasma membrane and where glucose uptake in the fasting phase is mediated by GLUT1 glucose transporter. HK2 operates in situ at 40% – 60% saturation. Here it is increased glucose uptake and increased in situ activity of HK2 in the fasting phase driven by mass action effects of increased fasting glucose concentration that stimulates unscheduled glycolysis. This activates processes decreasing insulin-stimulated glucose uptake in the absorptive phase or peripheral insulin resistance. The key determinant of increased fasting plasma glucose or impaired fasting glucose is hepatic insulin resistance giving rise to hypersecretion of glucose from the liver during the fasting phase (Rabbani & Thornalley, Frontiers in Endocrinology, 14: 1268308, 2024).
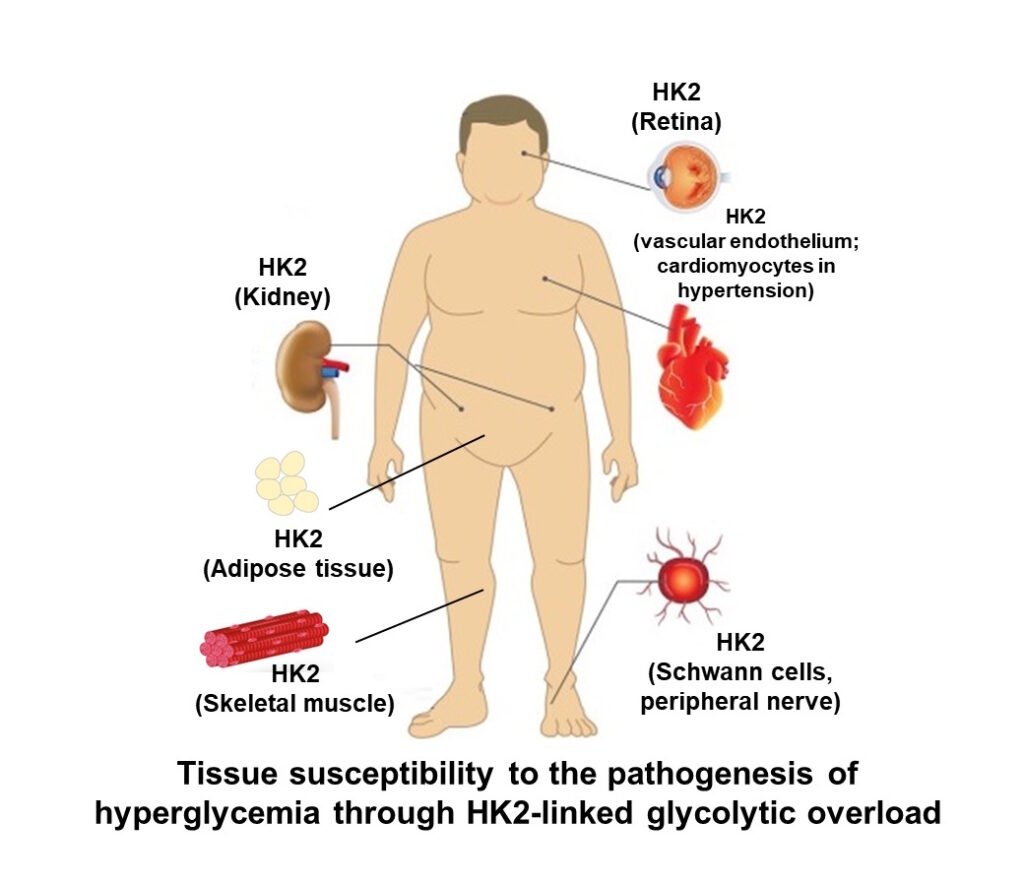
Paul J Thornalley 14th August 2025
Initiation of hexokinase-2 linked glycolytic overload – initiated by glucose-induced stabilization of hexokinase 2 to proteolysis
It is critical to know the mechanism of initiation of metabolic dysfunction in hyperglycemia as this is the key regulatory point to appreciate how increase and duration of increase in glucose concentration affects regulation of glucose metabolism. The initiation step also has multiple downstream pathways and only by correcting the key initiating step can all of the multiple downstream pathways be alleviated too. The key initiating step is glucose-induced stabilization of hexokinase 2 (HK2) to proteolysis with consequent increased HK2 protein and total hexokinase activity
High glucose-induced, HK2-linked responses are:
- Stabilization of HK2 to proteolysis by high cellular glucose concentration
- Increased flux of glucose metabolism without increased activity of glycolytic enzymes → wave of increased glycolytic intermediates. “Unscheduled glycolysis”
- This stimulates hexosamine, PKC and dicarbonyl stress pathways
- Increased G-6-P → detachment of HK2 from mitochondria, mitochondrial membrane polarization and increased reactive oxygen species (ROS)
- HK2 detached from mitochondria produces increased glycogen synthesis by metabolic channeling
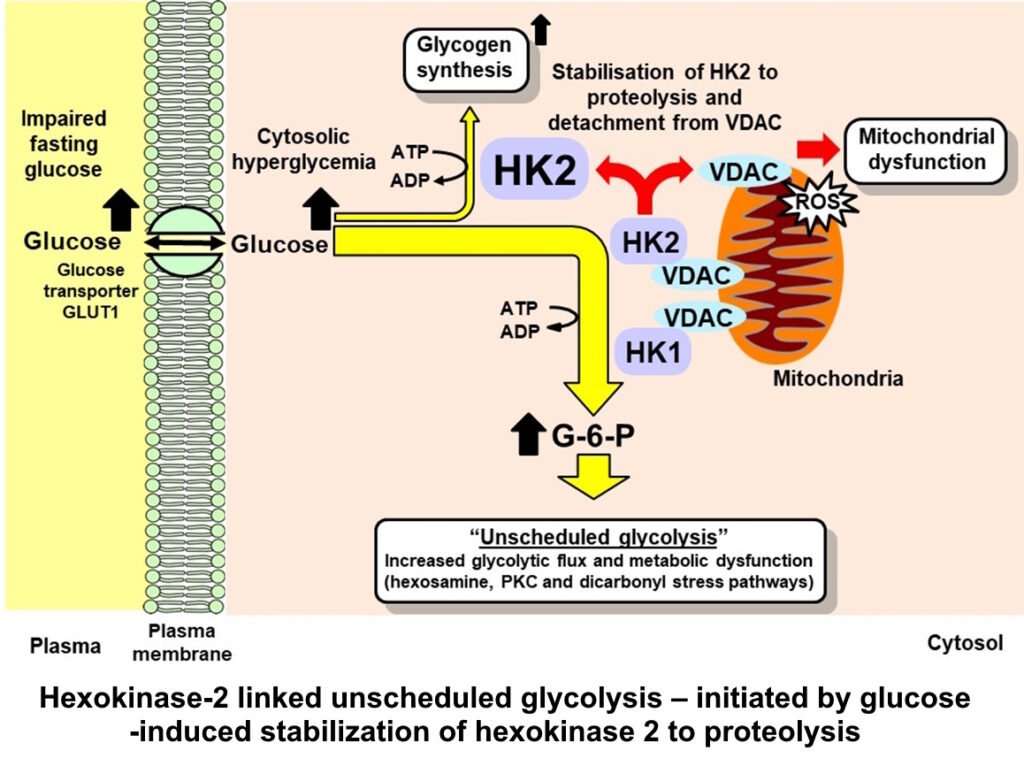
Abbreviations: G-6-P, glucose-6-phosphate; HK1 & HK2, hexokinase-1 & -2; VDAC, voltage-dependent anion channel. References: Irshad et al., Sci Rep 9, 7889, 2019; Ashour et al., BMJ Open Diabetes Research & Care. 8, e001458, 2020; Rabbani & Thornalley Frontiers in Endocrinol 14, 1268308, 2024
Paul J Thornalley 15th August 2025
Scheduled and unscheduled glycolysis
Normally when there is increased rate of glucose metabolism in cells – such as in skeletal muscle in the absorptive phase, insulin and transcriptional signaling through Mondo A increase expression and and activity of glycolytic enzymes, enhancing the onward metabolism of glycolytic intermediates and keeping the steady-state concentrations within the normal range. This is scheduled glycolysis. When increased flux of glucose metabolism occurs by increased hexokinase activity without increase in other early-stage glycolytic enzymes – glucose-6-phosphate isomerase (GPI), phosphofructokinase (PFK), aldolase (ALDO), triosephosphate isomerase (TPI) and glyceraldehyde-3-phosphate dehydrogenase (GA3PD), concentrations of glycolytic intermediates increase to abnormally high levels. Glucose-6-phosphate (G6P), fructose-6-phosphate (F6P), fructose-1,6-bisphosphate (F16BP), dihydroxyacetonephosphate (DHAP) and glyceraldehyde-3-phosphate (GA3P) all increase. This is unscheduled glycolysis. It occurs because for enzymes operating below saturation, flux v ≈ kcat/KM x [Enzyme][Substrate]. So, with [Enzyme], kcat and KM all unchanged, when flux v increases so does the steady-state substrate concentration [Substrate] correspondingly. For abnormally increased G6P, F6P, DHAP and GA3P, pathogenic mechanisms are activated.
I made a mathematical model of early-stage glycolysis with normal fasting plasma glucose of 5mM (FPG5), high fasting plasma glucose of 20 mM with not provision for stabilization of hexokinase-2 (HK2) to proteolysis by the high glucose concentration (FPG20) and high fasting plasma glucose of 20 mM with stabilization of HK2 to proteolysis inducing unscheduled glycolysis (FPG20_UG). Only in the latter case with increased HK2, glycolytic intermediates increased in high fasting plasma glucose – shown for G6P, F6P and rate of formation of methylglyoxal (MG) which forms spontaneously at trace levels from DHAP and GA3P. This is the mechanistic origin of metabolic dysfunction in hyperglycemia (Rabbani & Thornalley, Frontiers in Endocrinology, 14: 1268308, 2024). Presented at EASD 2022.
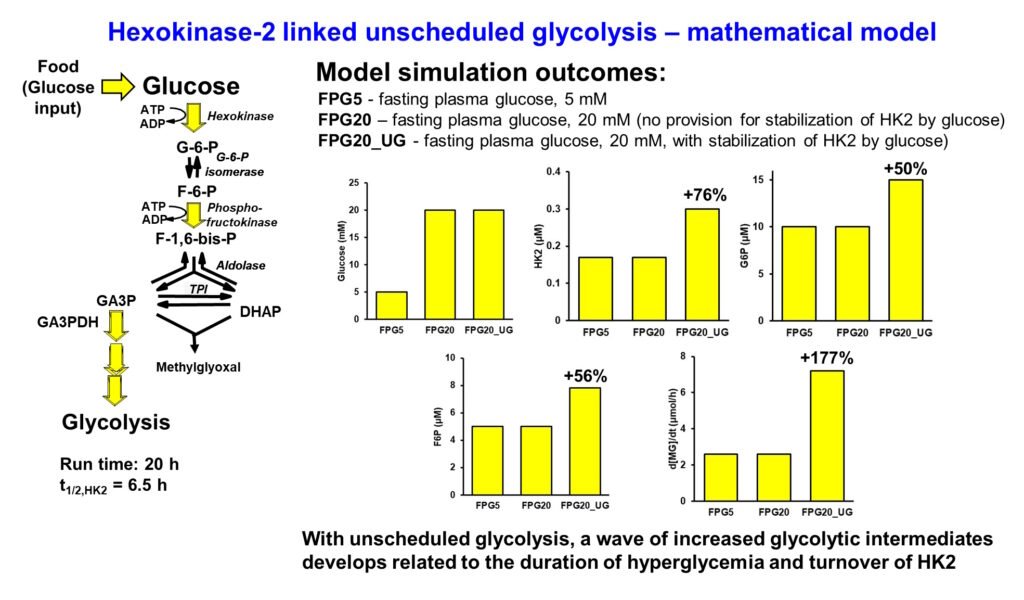
Paul J Thornalley 19th August 2025
Increased reactive oxygen species formation in glycolytic overload
The primary origin of increased reactive oxygen species (ROS) formation in glycolytic overload is detachment of hexokinase-2 (HK2) from mitochondria, leading to hyperpolarization of the mitochondrial membrane and increased formation of ROS from mitochondria. This was not recognized in the Oxidative stress hypothesis of 2001. The mitochondrial membrane becomes hyperpolarized on detachment of HK2 from binding to the voltage-dependent anion channel (VDAC) of the mitochondrial membrane because disposal of ATP from mitochondria through VDAC is about twice as fast when HK2 is attached to it compared to when HK2 is free in the cytosol. Other sources of increased ROS have been proposed: increased activation of NADPH oxidase through increased protein kinase C activity and inactivation of nitric oxide synthase. It is likely that these are secondary to increased formation of ROS from mitochondria.
A metabolic marker of detachment of HK2 from mitochondria is increased glycogen deposition as there is increased glycogen synthesis by metabolic channeling by the detached HK2. Increased glycogen can be found at sites of vascular complications of diabetes – kidney, retina and peripheral nerve; as also in early-stage embryogenesis in high glucose concentration (Rabbani & Thornalley, Frontiers in Endocrinology, 14: 1268308, 2024).
Increased ROS may induce oxidative stress and oxidative damage – for example decreased cellular thiols. In some cases, oxidative damage contributes to the pathogenesis e.g. diabetic embryopathy (reviewed in Wentzel et al. Antioxidants 2025, 14, 1022), and in some cases protein thiols are not decreased e.g. dermal fibroblasts in high glucose (Ashour et al., BMJ Open Diabetes Res Care 2020 8, e001458). It likely depends on antioxidant reserve in a particular cell type.
Finally, decreasing ROS alone does not counter all the effects of glycolytic overload in the pathogenesis of hyperglycemia. This is likely why antioxidants are not effective in the prevention and treatment of vascular complications of diabetes Rabbani & Thornalley, Frontiers in Endocrinology, 14: 1268308, 2024).
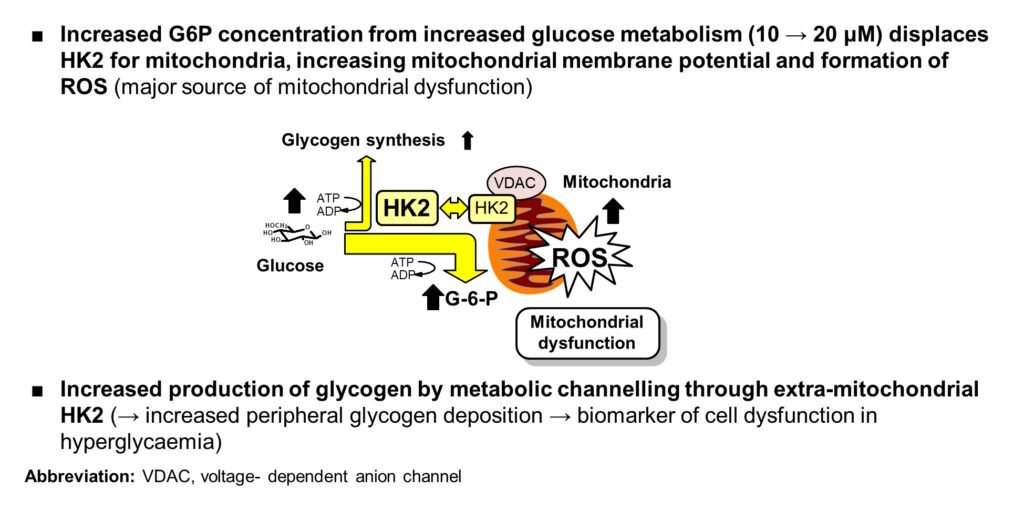
Origin of increased mitochondrial ROS in hyperglycemia (presented at EASD Meeting, Berlin, 2018)
Paul J Thornalley, 28th August 2025
Activation of the hexosamine pathway in glycolytic overload
The hexosamine pathway first came to prominence as a likely nutrient sensor mechanism linked to the development of peripheral insulin resistance, suggested by Rossetti and co-workers. Abnormally increased cellular concentrations of fructose-6-phosphate (F6P) increases the formation uridine-5′-di-phospho-N-acetyl-glucosamine (UDP-GlcNAc) and O-linked β-N-acetylglucosamine modification of serine and threonine residues of proteins or abnormally high enzymatic glycosylation. Targets for this modification are glucokinase, insulin receptor substrate proteins 1 and 2 (IRS-1 and IRS-2), forkhead transcription factor FoxO1, SP1, Mondo A, ChREBP, coat protein complexes (COPI and COPII), clathrin, soluble N-ethylmaleimide sensitive factor attachment protein receptors (SNAREs), and Golgi reassembly stacking protein of 55 kDa (GRASP55) that control protein trafficking and secretion, endothelial nitric oxide synthase (eNOS) and others. Increased glycosamine-6-phosphate of the hexosamine pathway is a competitive inhibitor of glucose-6-phosphate dehydrogenase and may thereby decrease pentose phosphate pathway activity supporting biosynthesis and antioxidant reserve. The hexosamine pathway is thereby involved in cell dysfunction in diabetes involved in insulin resistance, vascular complications and diabetic embryopathy.
Until the emergence of the glycolytic overload hypothesis, it was not clear how activation of the hexosamine pathway was initiated, and hence, how to prevent it for drug development. Now we know: by GlucoRegulate supplement which normalizes hexokinase-2, preventing glycolytic overload – including hexosamine pathway activation.
Impaired SP1-dependent expression of thiamine transporters through increased hexosamine pathway activity was implicated in the decreased renal expression of thiamine transporters linked to increased washout of thiamine in diabetes (Larkin et al, 2012).
Paul J Thornalley, 31st August 2025
Activation protein kinase c in glycolytic overload
It was suggested in 1989 that increased cellular concentrations of dihydroxyacetonephosphate (DHAP) in hyperglycemia gives rise to downstream increased glycerol-3-phosphate, lysophosphatidic acid, phosphatidic acid and diacylglycerol (DAG) – activator of protein kinase c (PKC). By this mechanism, there is abnormal activation of PKC was proposed to occur in hyperglycemia. The mechanism of increased DHAP was undisclosed but we now know this occurs in persistent hyperglycemia by hexokinase-2 linked glycolytic overload. The original paper on activation of PKC “Activation of protein kinase C by elevation of glucose concentration: Proposal for a mechanism in the development of diabetic vascular complications” was later retracted but subsequent studies have led to support for the abnormal activation of protein kinase c (PKC) involved in insulin resistance, vascular complications of diabetes and diabetic embryopathy. β- and δ-isoforms of PKC appeared activated preferentially in the vasculature in the diabetic state. Abnormal PKC activation led to increased production of extracellular matrix and cytokines, enhanced contractility, permeability, and vascular cell proliferation, activation of cytosolic phospholipase A2, and inhibition of Na+-K+-ATPase. Clinical studies followed – including with PKC inhibitor drugs, but likely as the mechanism of activation of PKC through glycolytic overload was unclear until recently, a safe and effective strategy to counter activation of PKC was not available. This is now available by treatment with GlucoRegulate.
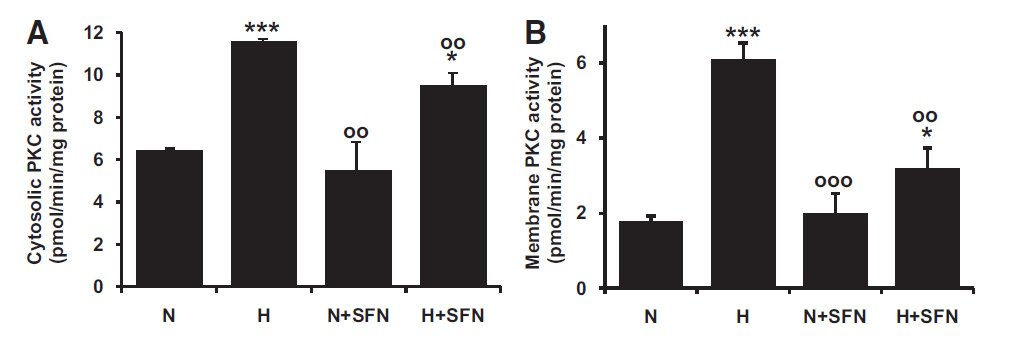
Activation of PKC in the microvascular endothelial HMEC-1 cell line by high glucose concentration in vitro. (Xue et al., Diabetes 57:2809–2817, 2008)
Paul J Thornalley 2nd September 2025
Increased formation of methylglyoxal, unfolded proteins and activation of the unfolded protein response (ER stress) in glycolytic overload
Increased triosephosphates, glyceraldehyde-3-phosphate (GA3P) and dihydroxyacetonephosphate (DHAP), in glycolytic overload increases the formation of reactive dicarbonyl metabolite, methylglyoxal (MG). My research established that subjects with diabetes have increased exposure to MG.
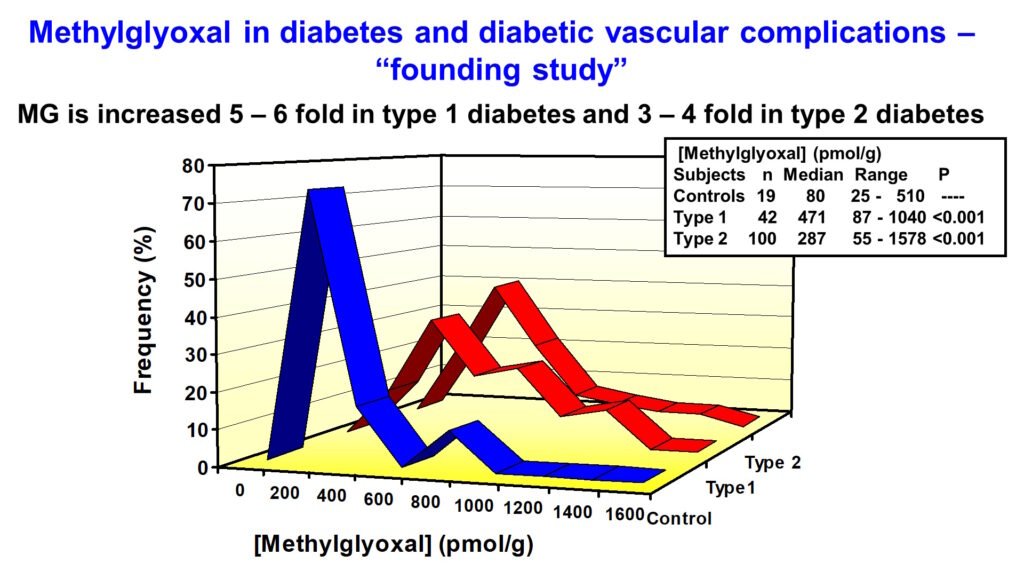
Abnormally increased MG is called dicarbonyl stress. MG is a potent glycating agent and precursor of the major physiological advanced glycation endproduct (AGE), MG-derived hydroimidazolone, MG-H1. Otherwise, MG is mainly metabolized by the glyoxalase system – see below. The cellular concentration of MG is increased due to both increased flux of formation of MG and decreased Glo1 activity linked to increased proteolysis of Glo1 protein (Irshad et al., Sci Rep 9, 7889, 2019).
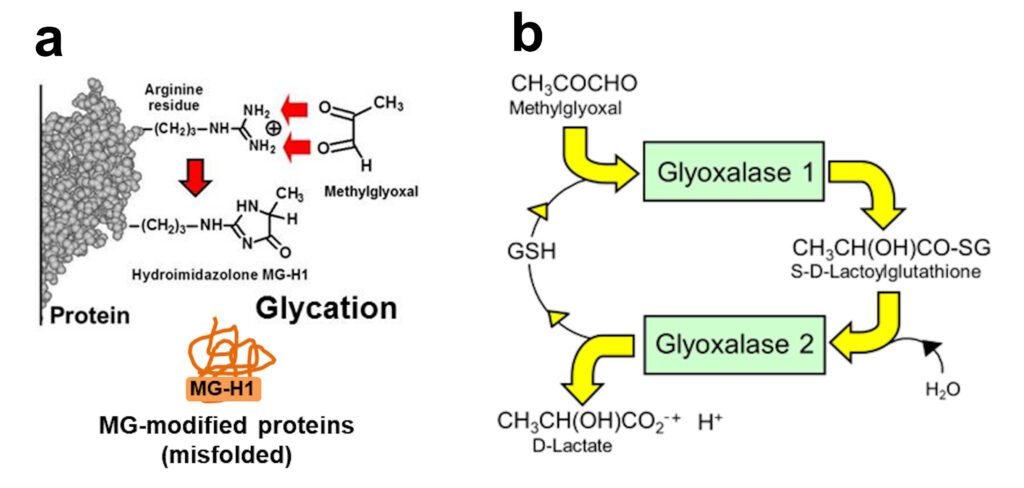
a. Glycation of arginine residues in proteins by methylglyoxal to form hydroimidazolone MG-H1 -misfolded MG-modified proteins. b. The glyoxalase system – major pathway of MG metabolism.
MG-modified proteins are major misfolded proteins in diabetes (Ahmed et al., Diabetologia 48, 1590–1603, 2005). Increased MG and MG-modified proteins activate the 3 endoplasmic reticulum-based sensors of the unfolded protein response: : inositol requiring enzyme-1α (IRE1α), double-stranded RNA-dependent kinase-like ER kinase (PERK) and activating transcription factor-6 (ATF6). Through this mechanism, increased MG – diccarbonyl stress – in hyperglycemia induces increased cellular proteolysis, apoptosis and low grade inflammation. This is prevented by GlucoRegulate.
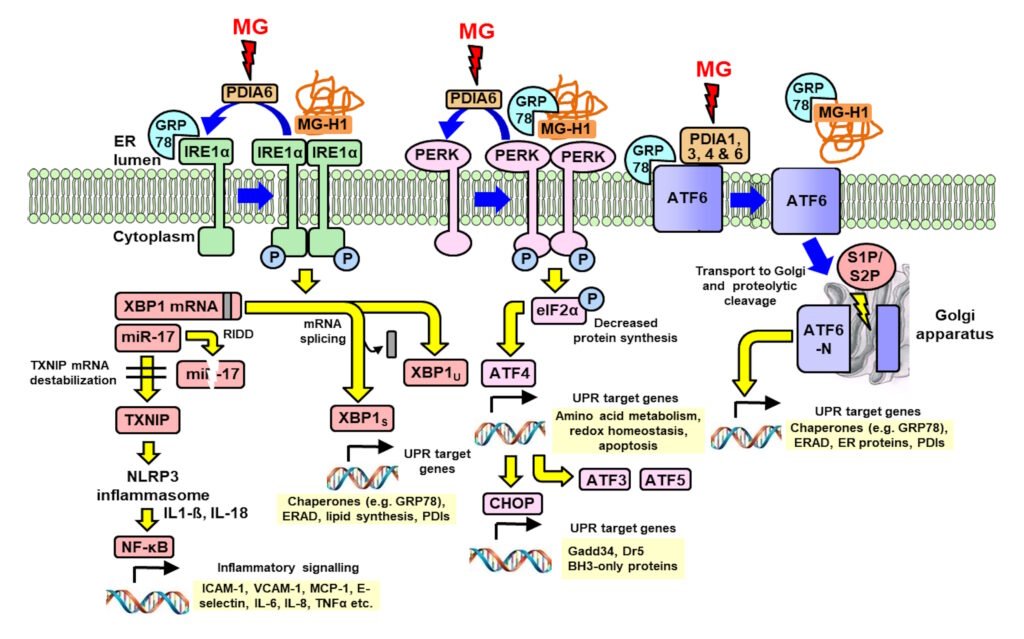
Activation of pathways of the UPR by interaction with methylglyoxal and methylglyoxal-modified proteins. Blue arrows are processes of UPR sensor activation and deactivation; yellow arrows are UPR signaling; and red arrows are protein disulfide isomerase (PDI) modifications by MG. Xue et al., Redox Biology 69, 103025, 1924.
Paul J Thornalley 3rd September 2025
Countering glycolytic overload with precision supplement, GlucoRegulate: preventing peripheral insulin resistance and vascular complications – induction of glucose-6-phosphate dehydrogenase
With regard to glycolytic overload driving the development of peripheral insulin resistance and vascular complications of diabetes, the most effective strategy for treatment is to decrease the activity of hexokinase-2 (HK2) to decrease the flux of glucose entering into glycolysis back to levels found in healthy control subjects. The expression of HK2 is regulated by glucose-6-phosphate (G6P)/Mondo A/Mlx transcriptional complex at peripheral and vascular sites. A strategy to do this is to induce expression of glucose-6-phosphate dehydrogenase (G6PD) to decrease cellular concentration of G6P and G6P/Mondo A/Mlx transcriptional activity. The G6PD gene has a functional antioxidant response element (ARE) in the promoter, so expression of G6PD can be increased by activating transcriptional factor nuclear factor erythroid 2-related factor 2 (Nrf2). This can be achieved safety and effectively with the optimized precision dietary supplement, GlucoRegulate – a combination of trans-resveratrol (tRES) and hesperetin (HESP). Increased expression of G6PD decreases cellular G6P concentration and thereby decreased G6P and G6P/Mondo A/Mlx transcriptional activity – countering HK2-linked glycolytic overload at course and also diverting increased metabolic flux into the pentosephosphate pathway for increased antioxidant reserve and biosynthesis by increased formation of NADPH.
For readers expert in the respiratory burst of neutrophils – I studied with the late, great scholar of neutrophils, Professor Filippo Rossi (University of Verona, Italy), it may be thought that the pentosephosphate pathway shunt will take increase G6P flux into the pentosephosphate pathway anyway as G6P concentration increases. Whilst this applies in neutrophils, it does not apply in vascular cells, muscle cells and adipocytes where the G6PD-catalysed forward reaction is rate-limiting and G6PD is saturated with G6P substrate even in normoglycemia. Increasing expression of G6PD is therefore required to increase the in situ rate of the G6PD-catalysed reaction.
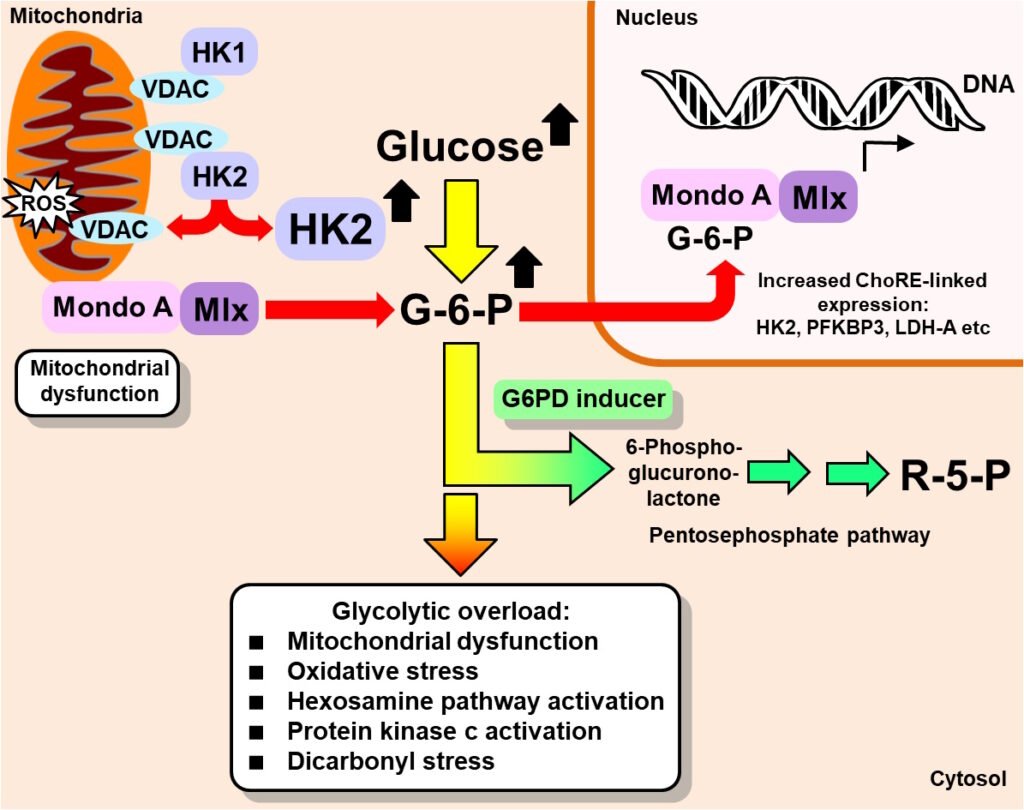
Induction of G6DP to counter hexokinase-2 linked glycolytic overload (Rabbani & Thornalley, Trends in Endocrinology & Metabolism 30, 419-431, 2019).
Paul J Thornalley, 26th September 2025
Countering glycolytic overload with precision supplement, GlucoRegulate: preventing peripheral insulin resistance and vascular complications – induction of glyoxalase 1
The reactive dicarbonyl methylglyoxal (MG) is a precursor of advanced glycation endproducts (AGEs) – long linked to the development of vascular complications of diabetes. Research findings of my team established: (i) MG concentration is increased in subjects living with obesity and patients with type 1 and type 2 diabetes; (ii) the major AGE-derived from MG is hydroimidazolone MG-H1 which increases at sites of development of vascular complications in experimental diabetes and is a risk predictor for the development of vascular complications in clinical diabetes; and (iii) increased MG and MG-H1-modified proteins – misfolded – are the major activators of the unfolded protein response or endoplasmic reticulum stress in hyperglycemia, supporting increased cellular proteolysis, apoptosis and low grade inflammation. Increased MG in hyperglycemia is a consequence of hexokinase-2 (HK2) linked glycolytic overload – not oxidative inhibition of glyceraldehyde-3-phosphate dehydrogenase, as had been proposed earlier. Increased formation of MG may be prevented by countering HK2-linked glycolytic overload and also by induction of expression of glyoxalase 1 (Glo1). Glo1 catalyzes the metabolism of MG by the cytosolic glyoxalase system – the major pathway of detoxification of MG. The precision dietary supplement, GlucoRegulate, was optimized for induction of expression of Glo1 through a functional regulatory antioxidant response element in the GLO1 gene. In double blind placebo-controlled crossover clinical trial, GlucoRegulate increased expression of Glo1, decreased MG and corrected insulin resistance in overweight and obese subjects. It also had a profound effect on low grade inflammation, decreasing monocyte chemoattract protein-1 (MCP-1), intereukin-8 (IL-8) and the receptor for advanced glycation endproducts (RAGE). Inhibitors of these inflammatory mechanisms has been sought for many years without success and now a precision dietary supplement hits all three! GlucoRegulate has recently been found to prevent diabetic embryopathy in an experimental model and now is a good supplement to treat insulin resistance and candidate for clinical evaluation to slow or prevent the development of endothelial dysfunction and vascular complications of diabetes.
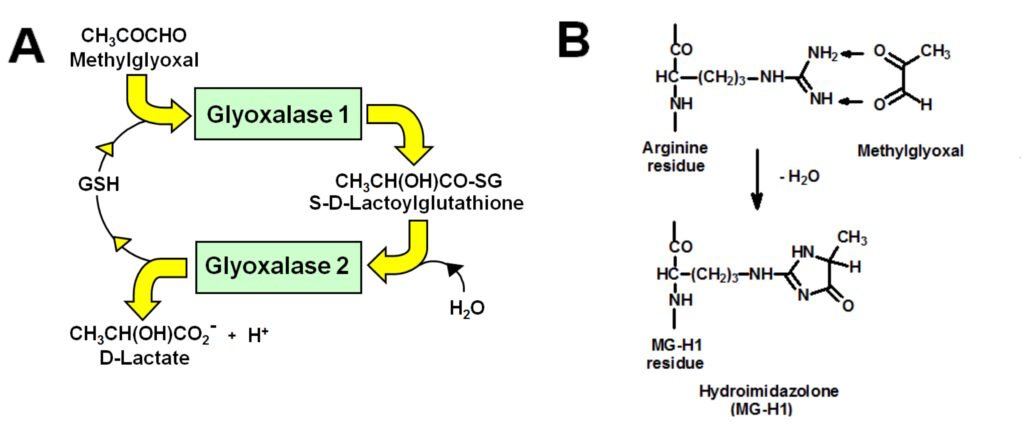
Figure. A. Metabolism of methylglyoxal by the glyoxalase system. B. Formation of major advanced glycation endproduct, MG-H1, from arginine residues by glycation with methylglyoxal.
Paul J Thornalley 28th September 2025
Countering glycolytic overload with precision supplement, GlucoRegulate: why earlier attempts to counter insulin resistance with resveratrol where unsuccessful
trans-Resveratrol has been evaluated in overweight and obese subjects in many clinical studies. Overall, in meta-analysis, there is no significant effect on plasma glucose and, where reported, insulin resistance at doses up to 1,500 mg per day – see Liu et al., Am J Clin Nutr 2014, 99, 1510 – 1519 and Batista-Jorge et al. Endocrines 2024, 5, 225 – 243. Despite this, the combination trans-resveratrol (90 mg) with hesperetin (120 mg) in a robust double blind, placebo-controlled crossover study in overweight and obese subjects improved fasting and postprandial glucose levels and insulin resistance – as assessed by the oral glucose insulin sensitivity (OGIS) index. See Xue et al., Diabetes 2016, 65, 2282 – 2294. We attribute this to improved bioavailability of trans-resveratrol in the presence of hesperetin and synergism of trans-resveratrol and hesperetin in activation of transcription factor Nrf2.
Activation of Nrf2 in the translocational oscillation model requires activation of sirtuin-1 to decrease inhibitory acetylation of Nrf2 in the cell nucleus and activation of fyn kinase to increase rate of export of Nrf2 from the nucleus for de-phosphorylation, de-acetylation and cellular stress status refresh in the cytosol.
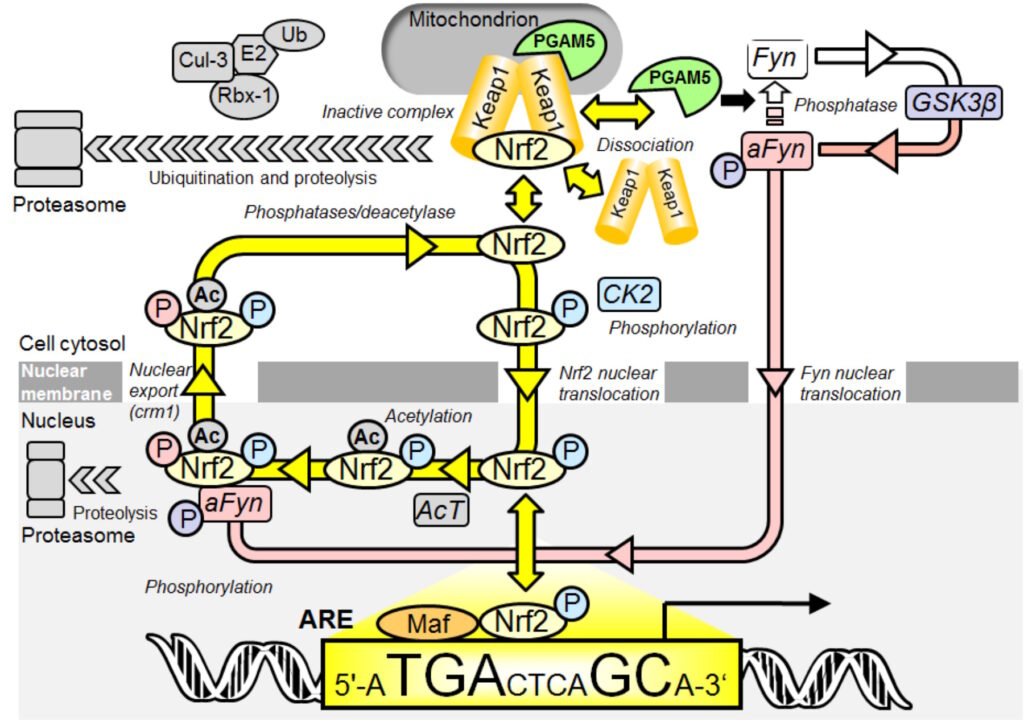
Figure: Oscillatory mechanism for Nrf2 regulation. Key: P, phosphorylation (color-coded for CK2 or Fyn catalyzed modification); and Ac, acetylation . In quiescence, Nrf2/Keap1/PGAM5 ternary complex in the cytoplasm is disrupted as Nrf2 is phosphorylated by CK2 and translocates to the nucleus to bind AREs with small MAF accessory proteins and activate basal ARE-linked gene expression. Therein Nrf2 becomes acetylated – causing functional inactivation. PGAM5 activates Fyn kinase which, after delay, translocates to the nucleus and further phosphorylates Nrf2 – driving nuclear expulsion. In the cytoplasm phosphorylations and acetylation are removed, stimulating complexation with Keap1 and PGAM5 which overshoots and the ternary complex fragments starting a further translocation cycle (yellow arrows). When activated with stimulants the translocation dynamics are enhanced. Nrf2 undergoes proteolysis in the cytoplasm and in the nucleus. We modelled ternary complex disruption mathematically in a generic form without discrimination for destabilisation mechanism. The model reproduces the key features of experimental observations: asymmetric oscillations and the faster oscillations of decreased amplitude when stimulated. A stochastic version of the model simulated using the Gillespie algorithm gives simulations that qualitatively match the experimental time course profiles and transcriptional response well. From Xue et al., Antioxid. Redox Signal. 23, 613–629.
trans-Resveratrol activates protein kinase A (PKA) by inhibition of phosphodiesterase-1 (PDE-1) and increase of cAMP. Hesperetin activates PKS directly. Synergistically activating PKA, sirtuin-1 and Fyn are both activated by phosphorylation by PKA. This activates Nrf2. Previously activation of Nrf2 was proposed to occur by inhibition of cellular proteolysis of Nrf2 and accumulation of Nrf2 protein. We showed, however, under dose-limiting conditions Nrf2 is activated without change in cellular concentration of Nrf2 proteins but rather by increased rate of translocation oscillation to and from the nucleus providing for increased refresh rate of Nrf2 when it returns inactivated to the cytosol.
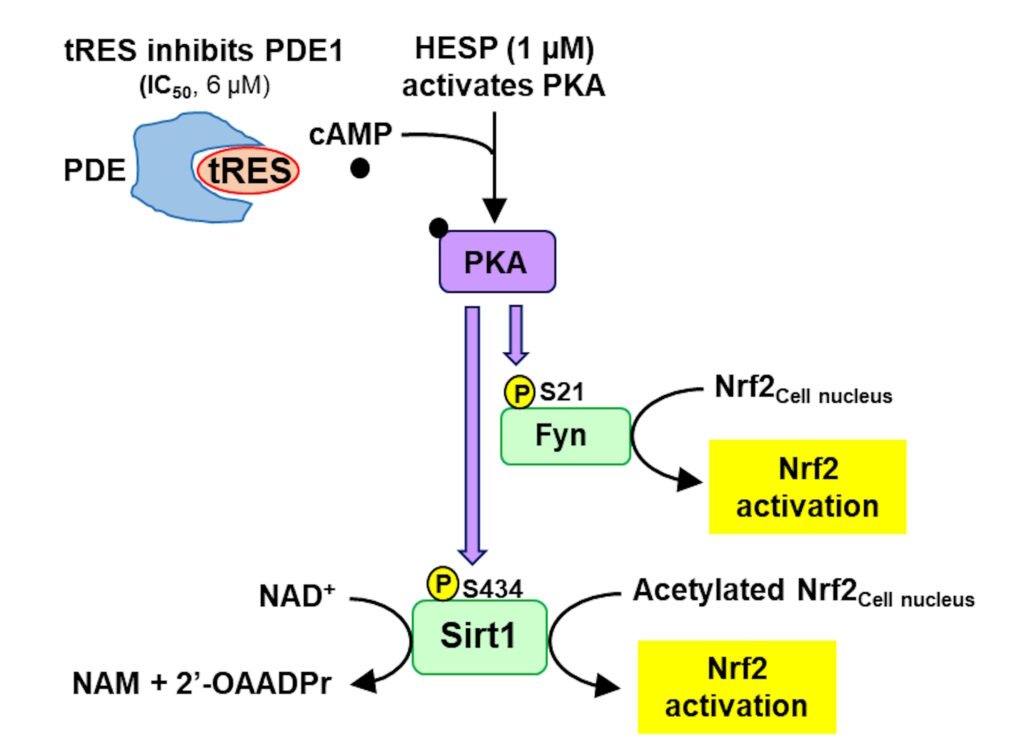
Figure: Pharmacological synergism of trans-resveratrol and hesperetin to activate sirtuin-1 and fyn kinase. See text. Abbreviations: PDE, phosphodiesterase; NAM, nicotinamide; 2’-OAADPr, 3′-O-acetyl-ADP-ribose; PKA, protein kinase A; Fyn, Fyn kinase; P, phosphorylation. From Xue et al., Antioxidants 2025, 14, 956
Paul J Thornalley 19th October 2025
Why might activators of Nrf2 failed in the past to deliver supplements and drugs to counter glycolytic overload, insulin resistance and complications of diabetes
The health benefits available through activation of transcription factor Nrf2 have long been known. It regulates the basal and inducible expression of ca. 1,300 genes with regulatory anti-oxidant response elements. Although research on Nrf2 is well-advanced, attempts to secure health benefits clinically from Nrf2 activators of botanical and synthetic origin have been disappointing and stalled. This is likely a consequence of incomplete understanding of how Nrf2 is activated, misunderstandings on the Nrf2 activator screening process, pursuing pharmacological agents activating Nrf2 at concentrations unlikely to be achieved clinically and insufficient attention given to the consideration that Nrf2 may be activated indirectly in response to toxicity with has problems relating to activator safety.
Initially, Nrf2 was considered to be activated by inhibition of the cellular proteolysis of Nrf2 proteins, with accumulation of Nrf2 being linked to Nrf2-linked transcriptional regulation. It was also claimed that Nrf2 activation does not show a normal dose-response curve and screening of activators was therefore done by assessing the amount of activators required to increase reporter response (e.g. ARE-linked luciferase reporter cell lines) over background by 2 or 10 fold. Unfortunately, these considerations, claims and approaches were not always applicable and often tended to be misleading. We showed, for example, that the well-known and studied activator of Nrf2 sulforaphane (SFN) activates Nrf2 at clinically relevant concentrations without change in cellular concentration of Nrf2 protein and it gave a typical dose-response curve – even with ARE-linked luciferase reporter assay (see figure below). The cellular accumulation of Nrf2 protein did occur but at higher concentrations of SFN which were not readily achievable clinically and also were associated with cytotoxicity.
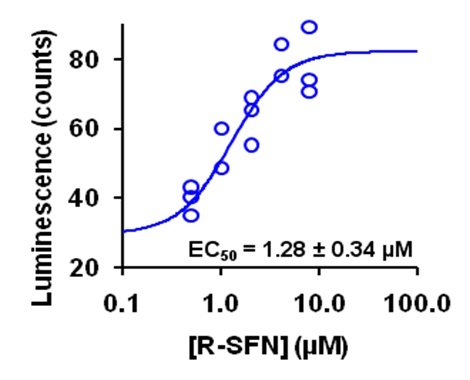
Figure: Dose response of NQO1-ARE transcriptional response on sulforaphane (SFN) concentration in HMEC-1 cells studied by luciferase reporter assay. Luminescence output is given in relative luminescence (counts). From Xue et al., Antioxid. Redox Signal. 23, 613–629.
We saw good dose response curves for trans-resveratrol (tRES) and cis-resveratrol (cRES) for induction of ARE-linked GLO1 expression. Crucially, we considered dietary bioactive compounds that were effective at clinically achievable concentrations and with very low toxicity – a high therapeutic index. The EC50 for trans-resveratrol was 2.5 μM with no cytotoxicity up to 40 μM (Xue et al., Diabetes 65, 2282–2294, 2016). The therapeutic index of synthetic activator of Nrf2, Bardoxolone methyl, was much lower: the EC50 for activation of Nrf2 was very low, ca. 25 nM, but the median growth inhibitory concentration GC50 was also very low, 110 nM, with overlapping response and cytotoxicity dose-response curves. This produces the suspicion that Nrf2 was being activated by Bardoxolone methyl in response to toxicity than engaging directly with the regulation of Nrf2. This may have contributed to later problems in clinical tolerability.
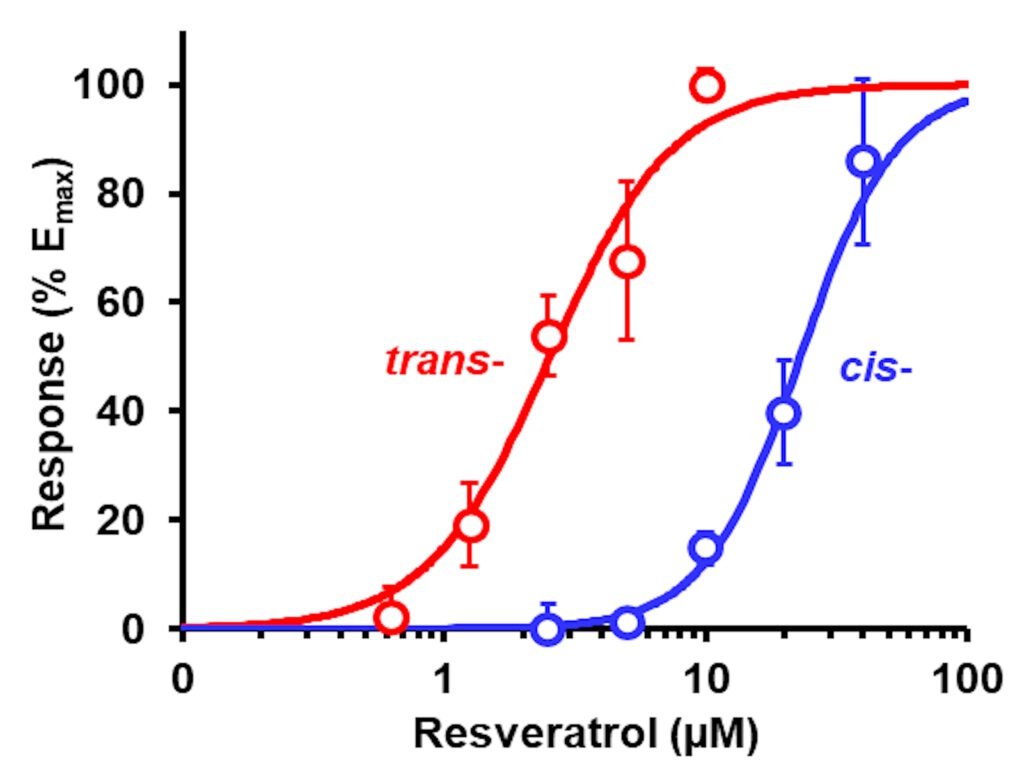
Figure: Induction of Glo1 expression by tRES and cRES. GLO1-ARE transcriptional response reporter assay. Data are mean ± SD (n = 3) for 5 concentrations. Nonlinear regression curves: tRES (red curve), E (%) = 100 × [tRES]1.99/(2.521.99 + [tRES]1.99); and cRES (red curve), E (%) = 100 × [cRES]2.36/(23.02.36+ [cRES]2.36). (Xue et al., Antioxidants
14, 956, 2025).
Paul J Thornalley, 25th October 2025
Countering glycolytic overload with precision supplement, GlucoRegulate: achieving an effective concentration of trans-resveratrol and hesperetin clinically for pharmacological activity
A major issue with clinical translation of health benefits of trans-resveratrol (tRES) has been pharmacological activity at concentrations which can be achieved clinically. Plasma concentrations of tRES maximize at the low micromolar level. In a recent paper, we considered the concentration dependence of the pharmacological responses to tRES, hesperetin (HESP) and tRES+HESP together in GlucoRegulate – see Table below. Clinically achievable responses are shown in green background; clinically unachievable responses in pastel pink background.

Table: Receptors for trans-resveratrol and hesperetin in cell responses. Key: green shading – effects at clinically achievable concentrations; pink shading -effects likely not clinically translatable.
The most potent response of tRES is activation of Nrf2. There may be also inhibition of phosphodiesterase 1 (PDE1) – likely contributing to activation of Nrf2, and antagonism at the aryl hydrocarbon receptor. Other effects observed in vitro at concentrations of tRES higher than achieved clinically are likely not operating clinically. For HESP, concentrations in plasma up to ca. 6 µM are easily achieved. So, activation of Nrf2, activation of protein kinase A – likely contributing to activation of Nrf2, and antagonism at the aryl hydrocarbon receptor likely come into play for pharmacological responses. When tRES is in the presence of HESP, the median effective concentration EC50 for tRES activation of Nrf2 decreases even further to 1.5 µM. There appears to be a plasma concentration range of ca. 1 – 5 μM, a clinical pharmacological “sweet spot,” where improvements in metabolic and vascular health are available. This has likely been achieved with GlucoRegulate where HESP enhances the effectiveness of tRES in activation of Nrf2. Xue, et al. Glyoxalase 1 Inducer, trans-Resveratrol and Hesperetin– Dietary Supplement with Multi-Modal Health Benefits. Antioxidants 14, 956, 2025. With GlucoRegulate,, therefore, we now have safe and clinically effective activator of Nrf2 to achieve related health benefits.
Paul J Thornalley, 1st November 2025
Where decline in glycemic health starts for most people: hepatic insulin resistance
Diets rich in simple sugars lead to overactivation of hepatic carbohydrate response element binding protein (ChREBP) which synergizes with fats to down regulate expression of insulin receptor substrate-2 (IRS-2) to produce hepatic insulin resistance, increasing FPG. There is also down regulation of IRS-2 expression by insulin-dependent IRS-1 signalling in hyperinsulinemia – explaining why hyperinsulinemia may precede hepatic insulin resistance in some people with hypersecretion of insulin. IRS-2 expression is also decreased by IRS-1 linked signalling through sterol response element binding protein-1c (SREBP1c), potentiated by lipid-linked oxysterol-dependent activation of liver X
receptor-α (LXRα) for maximum effect.
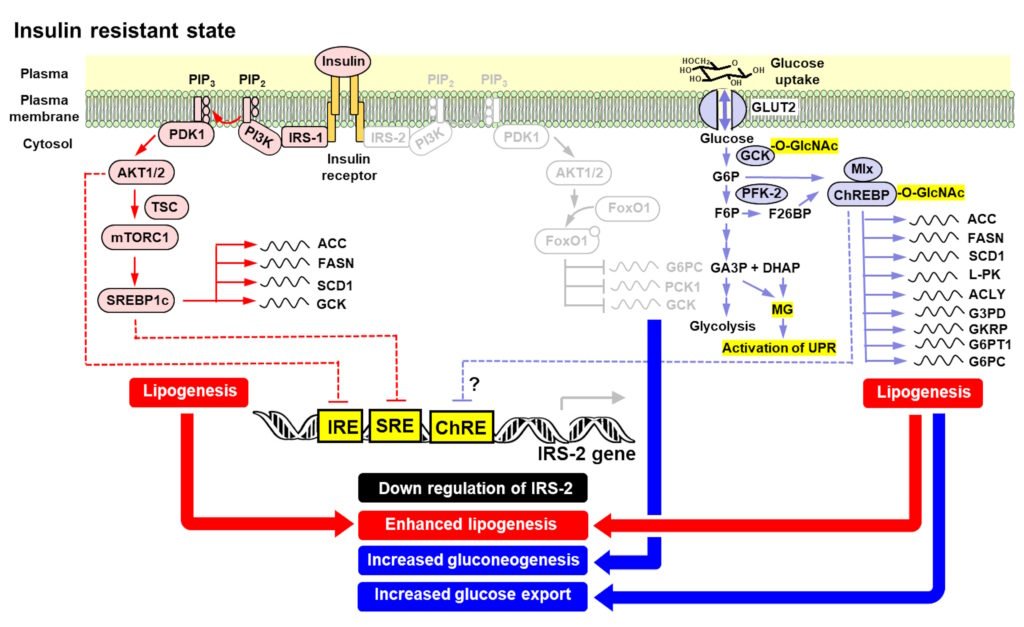
Figure: Mechanisms of hepatic insulin resistance, de novo lipogenesis and increased gluconeogenesis increasing fasting plasma glucose on a sugar-rich diet. From: Rabbani and Thornalley Clin Sci 139, 1405–1429, 2025.
With expression of IRS-2 decreased, the liver is continually producing glucose and then is a risk of glycolytic overload through excessive glucose production and metabolism. To avoid this, ChREBP increases expression of glucose exporting genes: G6P translocase (G6PT1) – which transports G6P from the cytosol to the lumen of the ER where glucose-6-phosphatase (G6PC) residues; and G6PC which converts G6p to glucose. The liver then switches to continual hyper-secretion of glucose. This increases fasting plasma glucose concentration, exposing skeletal muscle, adipose tissue and pancreatic beta-cells to continual increased glucose exposure. This is the start of hyperglycemia-induced peripheral insulin resistance and beta-cell glucotoxicity – the latter leading to increased secretion of insulin and stress of beta-cells and their down regulation of glucokinase and increased expression of hexokinases -1 and -2 as dedifferentiation of beta-cells begins on the path to decline of insulin secretion response and type 2 diabetes.
GlucoRegulate offers a new treatment for hepatic insulin resistance. Through activation of Nrf2, it increases the expression of glucose-6-phosphate dehydrogenase (G6PD), thereby decreasing glucose-6-phosphate (G6P) by diverting increased flux of it to the pentosephosphate pathway. This decreases activation of G6P/ChREBP/Mlx transcriptional signalling downregulation IRS-2, re-instating hepatic insulin sensitivity. It also decreases expression of sterol response element binding protein-1c (SREBP1c) which also counters decreased expression of IRS-2 and decreases hepatic lipogenesis. In the HATFF clinical evaluation of GlucoRegulate, insulin resistance was corrected which includes hepatic insulin resistance. See Xue et al., Diabetes 65, 2282–2294, 2016. We describe hepatic signalling in response to sugar rich diets in our recent paper – Rabbani and Thornalley Clin Sci 139, 1405–1429, 2025.
Paul J Thornalley, 21st November 2025