Summary
Hyperglycemia is a risk factor for the development of insulin resistance, beta-cell glucotoxicity, and vascular complications of diabetes. We propose the hypothesis, hexokinase-linked glycolytic overload and unscheduled glycolysis, in explanation. Hexokinases (HKs) catalyze the first step of glucose metabolism. Increased flux of glucose metabolism through glycolysis gated by HKs, when occurring without concomitant increased activity of glycolytic enzymes—unscheduled glycolysis—produces increased levels of glycolytic intermediates with overspill into effector pathways of cell dysfunction and pathogenesis. HK1 is saturated with glucose in euglycemia and, where it is the major HK, provides for basal glycolytic flux without glycolytic overload. HK2 has similar saturation characteristics, except that, in persistent hyperglycemia, it is stabilized to proteolysis by high intracellular glucose concentration, increasing HK activity and initiating glycolytic overload and unscheduled glycolysis. This drives the development of vascular complications of diabetes. Similar HK2-linked unscheduled glycolysis in skeletal muscle and adipose tissue in impaired fasting glucose drives the development of peripheral insulin resistance. Glucokinase (GCK or HK4)-linked glycolytic overload and unscheduled glycolysis occurs in persistent hyperglycemia in hepatocytes and beta-cells, contributing to hepatic insulin resistance and beta-cell glucotoxicity, leading to the development of type 2 diabetes. Downstream effector pathways of HK-linked unscheduled glycolysis are mitochondrial dysfunction and increased reactive oxygen species (ROS) formation; activation of hexosamine, protein kinase c, and dicarbonyl stress pathways; and increased Mlx/Mondo A signaling. Mitochondrial dysfunction and increased ROS was proposed as the initiator of metabolic dysfunction in hyperglycemia, but it is rather one of the multiple downstream effector pathways. Correction of HK2 dysregulation is proposed as a novel therapeutic target. Pharmacotherapy addressing it corrected insulin resistance in overweight and obese subjects in clinical trial. Overall, the damaging effects of hyperglycemia are a consequence of HK-gated increased flux of glucose metabolism without increased glycolytic enzyme activities to accommodate it.
Hexokinase-linked glycolytic overload and unscheduled glycolysis in hyperglycemia-induced pathogenesis of insulin resistance, beta-cell glucotoxicity, and diabetic vascular complications
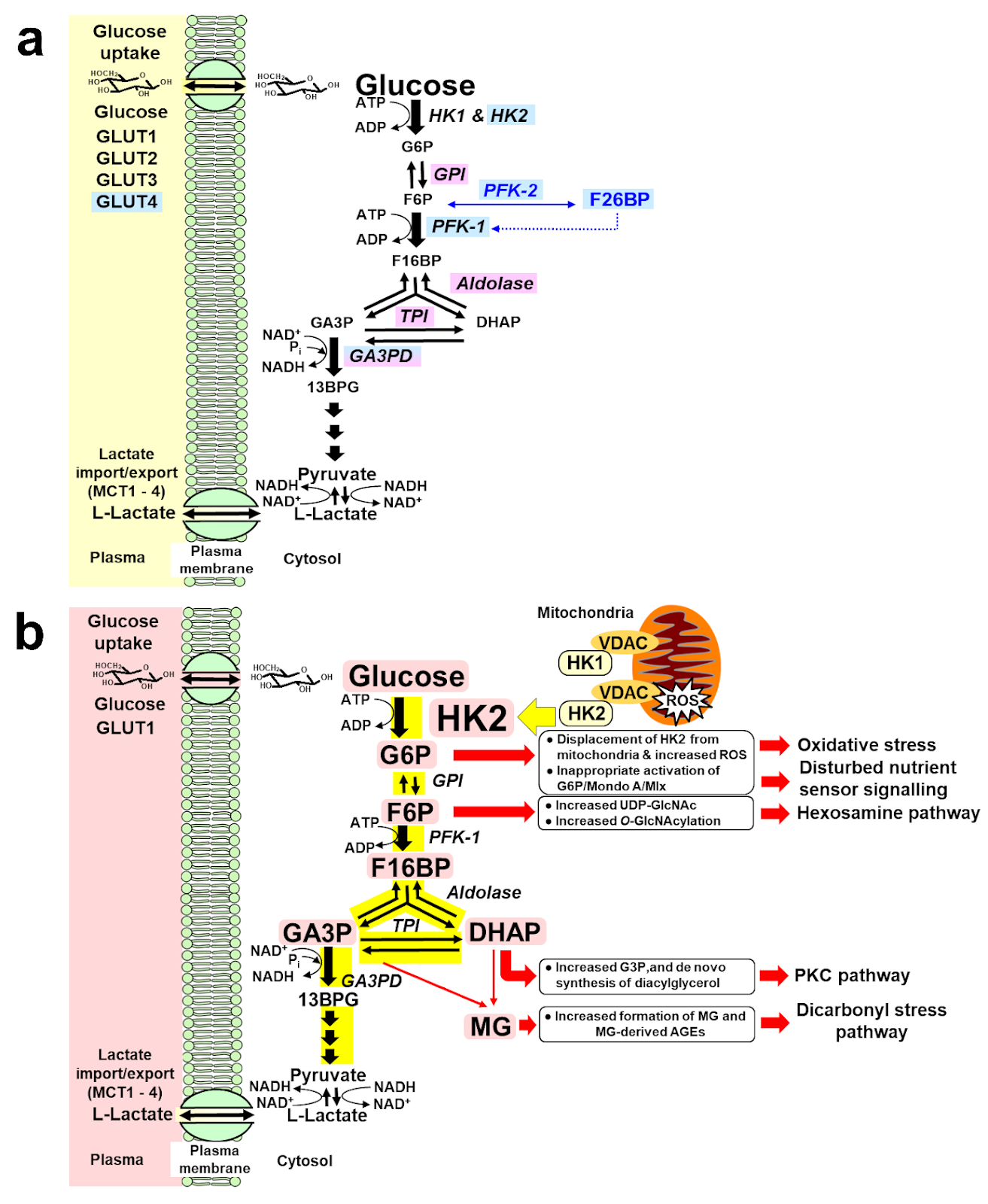
Figure 1. a. Regulation of glycolytic enzymes by insulin and Mondo A in scheduled glycolysis. Key: blue highlighted text, genes with expression and/or activity regulated by insulin. Lilac highlighted text, genes with expression regulated by Mondo A/Mlx. b. Dysregulation of glycolytic enzymes and metabolic dysfunction in hexokinase-2 linked glycolytic overload and unscheduled glycolysis hypothesis. Key: Pink highlighted text – HK2 and metabolites increased in unscheduled glycolysis.
Principal publication
Unraveling the impaired incretin effect in obesity and type 2 diabetes: Key role of hyperglycemia-induced unscheduled glycolysis and glycolytic overload
Glucagon-like peptide-1 (GLP-1) agonists and GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) co-agonists are major treatment options for subjects with obesity and patients with type 2 diabetes mellitus (T2DM). They counter without addressing the mechanistic cause of the impaired incretin effect associated with obesity and T2DM. Incretin effect impairment is characterized by decreased secretion of incretins from enteroendocrine cells and incretin resistance of pancreatic β-cells. It is linked to hyperglycemia. We present evidence that subversion of the gating of glucose entry into glycolysis, mainly by glucokinase (hexokinase-4), during persistent hyperglycemia in enteroendocrine cells, pancreatic β- and α-cells and appetite-regulating neurons contributes to the biochemical mechanism of the impaired incretin effect. Unscheduled glycolysis and glycolytic overload thereby produced decreases cell signalling of incretin secretion to glucose and other secretion stimuli and incretin receptor responses. This mechanism provides a guide for development of alternative therapies targeting recovery of the impaired incretin effect.
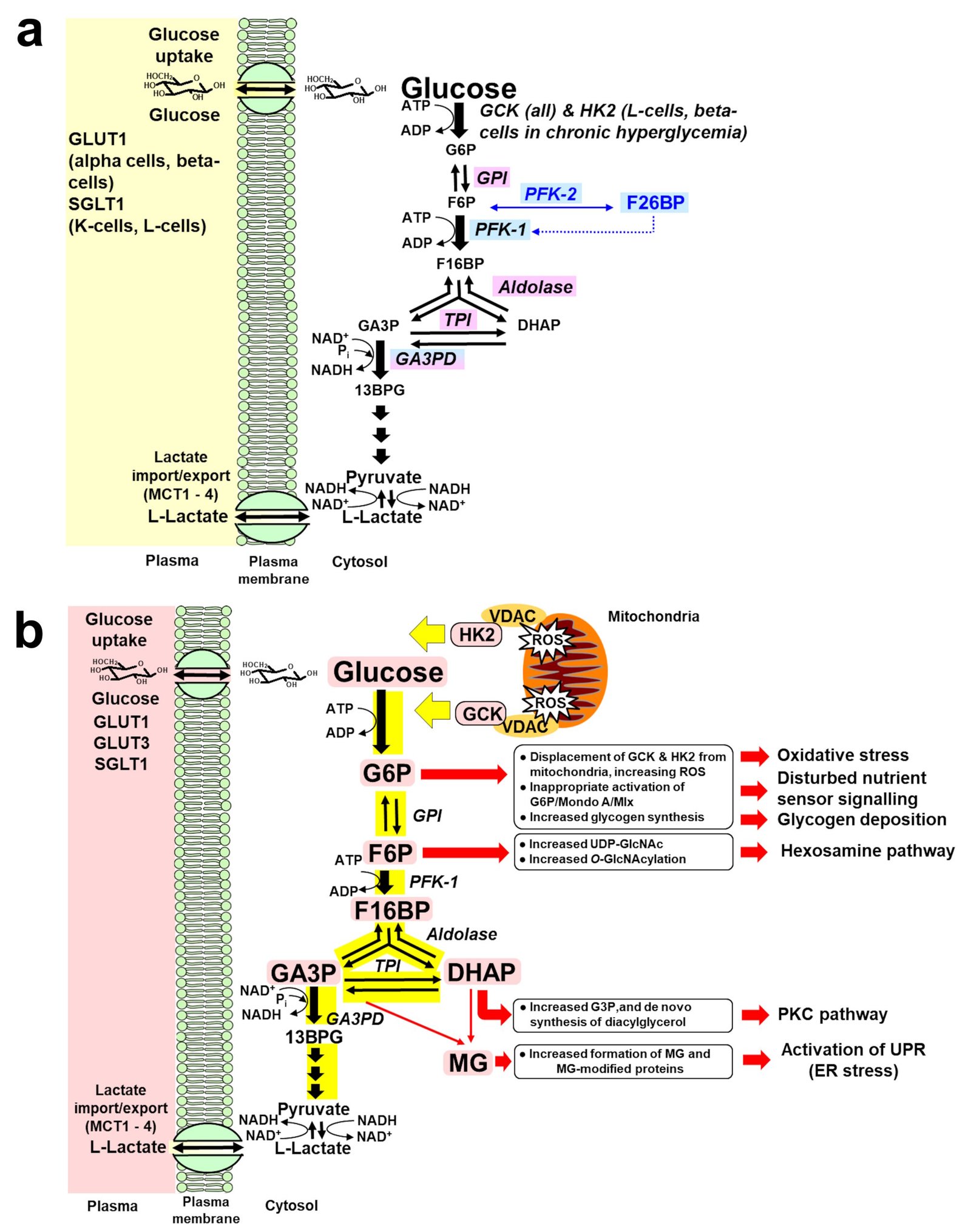
Figure 2. a. Regulation of glycolytic enzymes by insulin and Mondo A in scheduled glycolysis. Key: blue highlighted text, genes with expression and/or activity regulated by insulin. Lilac highlighted text, genes with expression regulated by Mondo A/Mlx. b. Dysregulation of glycolytic enzymes and metabolic dysfunction in GCK and HK2-linked glycolytic overload and unscheduled glycolysis hypothesis. Key: Pink highlighted text – HK2 and metabolites increased in unscheduled glycolysis. For GCK, detachment from VDAC also promotes oligomerization of Bax, cytochrome c release and apoptosis [120]. Abbreviations: DHAP, dihydroxyacetone-phosphate; F-6-P, fructose-6-phosphate; F-1,6-bis-P, fructose-1,6-bisphosphate; F-2,6-bis-P, fructose-2,6-bisphosphate; G-6-P, glucose-6-phosphate; GA3P, glyceraldehyde-3-phosphate; MCT, monocarboxylate transporter; ROS, reactive oxygen species; VDAC, voltage-dependent anion channel.
Principal publication